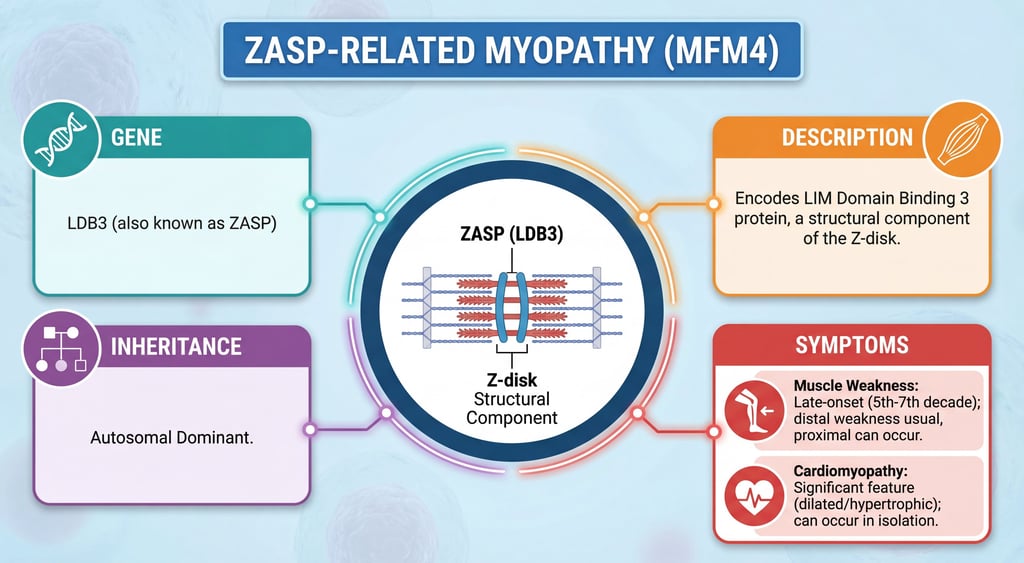
Also known as Zasp or Zaspopathy
Autosomal dominant inheritance
A form of myofibrillar myopathy, a group of chronic neuromuscular disorders characterized at ultrastructural level by disintegration of the sarcomeric Z disk and myofibrils, and replacement of the normal myofibrillar markings by small dense granules, or larger hyaline masses, or amorphous material. MFM4 is characterized by distal and proximal muscle weakness with signs of cardiomyopathy and neuropathy. Protein name - LIM domain-binding protein 3 Amino Acid change Serine/Threonine
A DNA test result may look like this: c.439 G>A; p.Ala147Thr c.429G>A p.Ala147Thr c.494C>T p.Ala165Val c.76C>T p.Pro26Ser
The four different missense mutations in the LDB3 gene (A147T, A165V, R268C and P26S ), were found in patients in a heterozygous form, causing myofibrillar myopathy. The first two mutations occurred in exon 6, and R268C occurred in exon 9 and P26S on exon 1. Since all mutations were detected on the same gene in LDB3, a similar phenotype might be expected in the patients of both families such as progressive proximal and/or distal weakness, cardiac involvement and peripheral neuropathy. Mutant LIM domain-binding protein 3 (ZASP) is predicted to weaken the linkage of Z-disk filaments to thin filaments. This novel mutation (c.1687A > G (p.Ile563Val) mutation in exon 10) may change the function of the ZASP-L and LIM domains.


Stories from people living with LDB3
US WI male 1
Unknown to me at the time I started experiencing pain in my calves and lower back when I was in my mid-20s. I was able to relieve the pain by wearing work boots with high heels or have my heels raised. My father also had to use this remedy but he did not MFM. My MFM came from my mother’s side.
After I turn 45 I noticed my energy level was dropping and shortly after that I developed a problem in my right hip which was causing me to limp. I went to therapy since the doctors couldn’t find the problem but I only had a slight improvement. After I was 50 I learned my brother was having similar issues only his weakness came on very suddenly where is mine progressed very slowly. One of his doctors said he might have a form of muscular dystrophy but my brother never pursued a complete diagnosis.
I went through 3 doctors before the 4th one gave me a complete diagnosis of MFM4 Ala147Thr - which took about 5 years. I had two ELECTROMYOGRAPHY (EMG) test, one MUSCLE BIOPSY from the upper arm and my highest CREATINE KINASE test was near 1600. Since then it continues to progress so at 65 years old I am completely wheelchair-bound since my legs cannot support me to even stand. My arms are also extremely weak to the point I have to have my face 6 inches from my plate to eat. My fingers are extremely rigid and I am unable to make a fist so it is very difficult to pick up things of any weight. I can still drink out of a plastic cup by squeezing it between my pointer and middle finger.
I have had to modify my computer station with a standing desk to raise up to fit my power wheelchair under. I have purchased voice recognition software so I can type and I’ve also purchased a device to move my mouse with head motions but that system did not work well. It makes me happy that I can still mow my lawn but now I have to have someone lift me up using a hoist and sling to place me into the seat.
In the past I have tried different supplements but did not notice any improvements in my condition. I do not have any other medical issues except high cholesterol which is only getting worse because I can’t do any physical activity. I do not have any issues with my heart or lungs at this time. I take Gabapentin for muscle pain and I have to keep myself warm, my legs are very cold all the time.
Update at 69: Still getting weaker and my fingers are becoming more useless as all the muscles are turning into fatty tissue. I use one finger to type now when I cannot use Microsoft 365 dictate button in Word. Always very cold and wear a electric throw blanket most of the time.
US MN female 1
Back in the 1970's, my father and his sister were determined to figure out what in the world they had. All they knew is that it was "what happened to Grandma." Other family members were stricken also, but many of them died before they showed the most severe symptoms. This seems to be a common thread among many sufferers, an inability to receive a diagnosis, and some horrendous misdiagnoses and subsequent traumatic, futile “treatment.”
My father died at 83 in 2005, bed-bound and unable to even push a button for help. His diagnosis had settled by then on “perhaps some type of distal dystrophy.” He would have been amazed to know that we have an identifiable genetic flaw, LBD3, MFM4. I'm one of six of his children: three are symptomatic and three are fine.
I'm 67 and still walking, though that's getting to be a real challenge. With walking sticks I manage. My sister is 66 and about the same. My heart's OK, and my diaphragm. My brother is 54 and still doing fine, but he had the blood test that shows he has the genetic defect, and there are ways he can tell. For us, trouble climbing stairs are the first indication.
We are so lucky compared to some. I take two ibuprofen daily just because it makes my gait feel a bit more normal. I walk with hiking poles for balance. I am taking CoQ10 (recommended dose on the bottle) but I don't know if it's making a difference. My sister takes Celebrex (generic version) and it makes her legs less uncomfortable. My brother takes CoQ10.
I'm planning to start taking NAC (N-Acetyl Cysteine) soon and possibly creatine. I feel that while there is no cure right now, there may be possible treatments that can affect the progress of the muscle deterioration.
I'm certainly willing to speak to anyone who might be interested in my experiences or family history. I live in Minnesota, my sister lives in New Mexico, and my brother in Denver.
US MI male 1
My dad was having issues and was diagnosed with LGMD about the time I was born. I grew up watching him get worse. Of course in the 1960’s they didn’t know what this was or what caused it. They told him “we believe an enzyme is eating away the muscle.”
My dad was first in our family’s history to have this indicating a dominant de novo mutation. My two sisters were spared.
I was a slow-to-normal kid. I could run, jump, play ball, etc. but was always slower than my friends and schoolmates. I dreaded gym class. In 1973, I was 12. My dad and I were admitted to an MD Clinic hospital with other patients for a weekend long evaluation. This included observation, EMGs and an open biopsy. My tests all came back inconclusive with no smoking-gun evidence I had MD but neither could they rule it out.
Around age 38 I knew in my heart something was wrong. I had difficulty rising from a sofa or the floor. People 75 years old could pass me on a stairway. I tripped often and sometimes went down. I hid it for 3 years, but finally went in and was diagnosed at age 41. The EMG and biopsy, this time, left no doubt.
Half a dozen hit-or-miss single gene tests came back negative. Later, several NMD gene panels didn’t give me an answer either. I became an advocate for obtaining a diagnosis. In April 2019, after having “some kind of LGMD” for 17 years, I received the long awaited call. Whole Exome Sequencing at Havard revealed a flaw in my LDB3 gene resulting in MFM4.
I began using a power chair full time at age 56, fifteen years after diagnosis. I’ve been spared cardiac involvement and I have minor breathing and swallowing issues. I take oral Albuterol, Vitamin D3 and CoQ10. I feel all have been beneficial.